| variant | sequences | date | |
|---|---|---|---|
| 0 | wildtype | 1 | 2022-01-01 |
| 1 | wildtype | 12 | 2022-01-02 |
| 2 | wildtype | 17 | 2022-01-03 |
| 3 | variant | 1 | 2022-01-03 |
| 4 | wildtype | 11 | 2022-01-04 |
| 5 | variant | 1 | 2022-01-04 |
| 6 | wildtype | 15 | 2022-01-05 |
From Sequencing to Surveillance: Estimating SARS-CoV-2 variant fitness
March 11, 2024
Evolution and selection in SARS-CoV-2
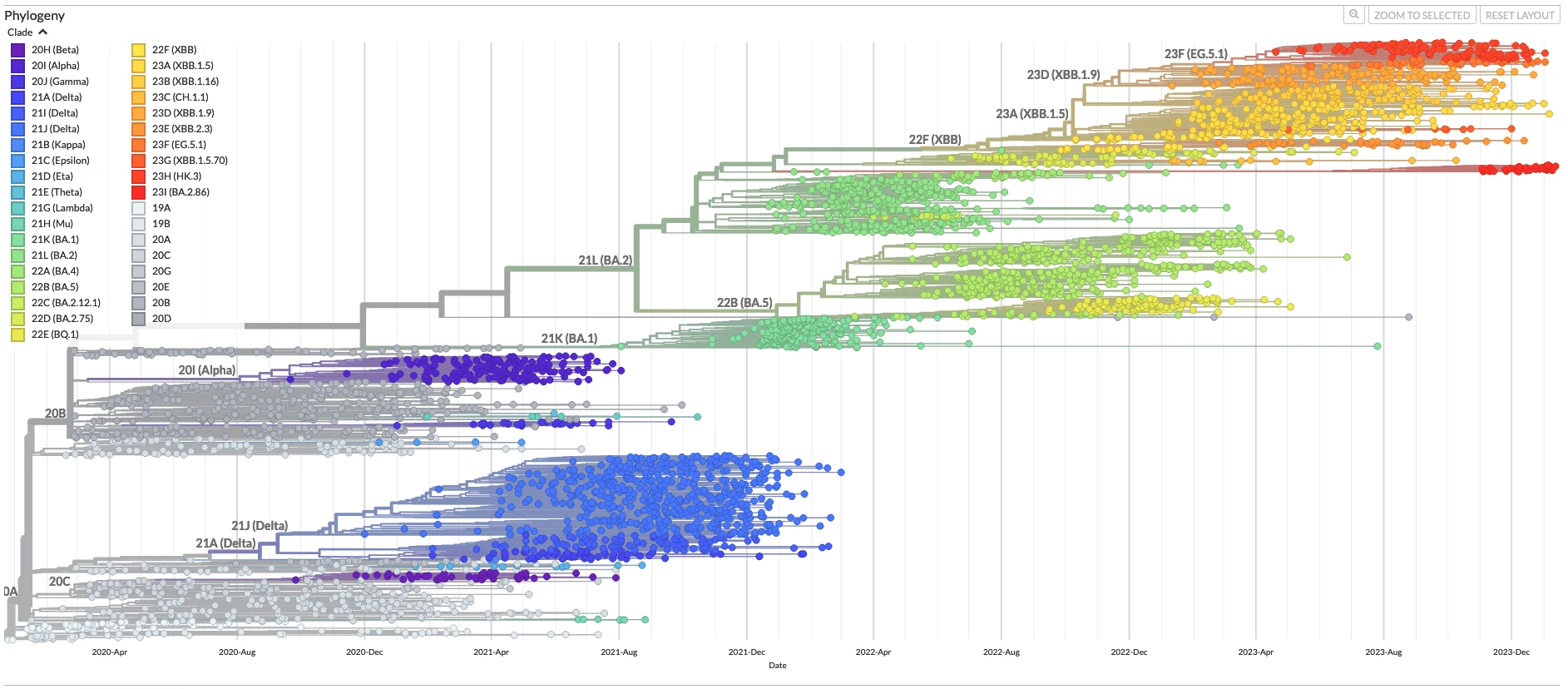
All-time phylogenetic tree for SARS-CoV-2 from Nextstrain
Selection and Evolution
- Selection is the process by which individuals have higher fitness in certain environments
- Evolution is the change in the genetic composition of the population over time due to selection and heritable variation
- Relative fitness is the relative capacity for individuals to reproduce in a population


Illustrating selection
![]()
- An early mutation that takes us from green infections to purple which cause more secondary infection.
- In this case, the purple is selected for.
Quantifying selection and relative fitness
- Often, we’re interested in not just the presence of selection but its magnitude.
- When quantifying selection, you’ll often see selective coefficient or relative fitness discussed.
- Relative fitness is the difference in the growth rates of two variants i.e. the relative capacity for individuals to reproduce in a population



Remark: For those familiar with the selective coefficient \(s\), the relative fitness \(\lambda\) is given by \((1 + s) = \exp(\lambda)\)
Generating sequence counts

Estimating relative fitness with evofr


Estimating relative fitness across multiple locations
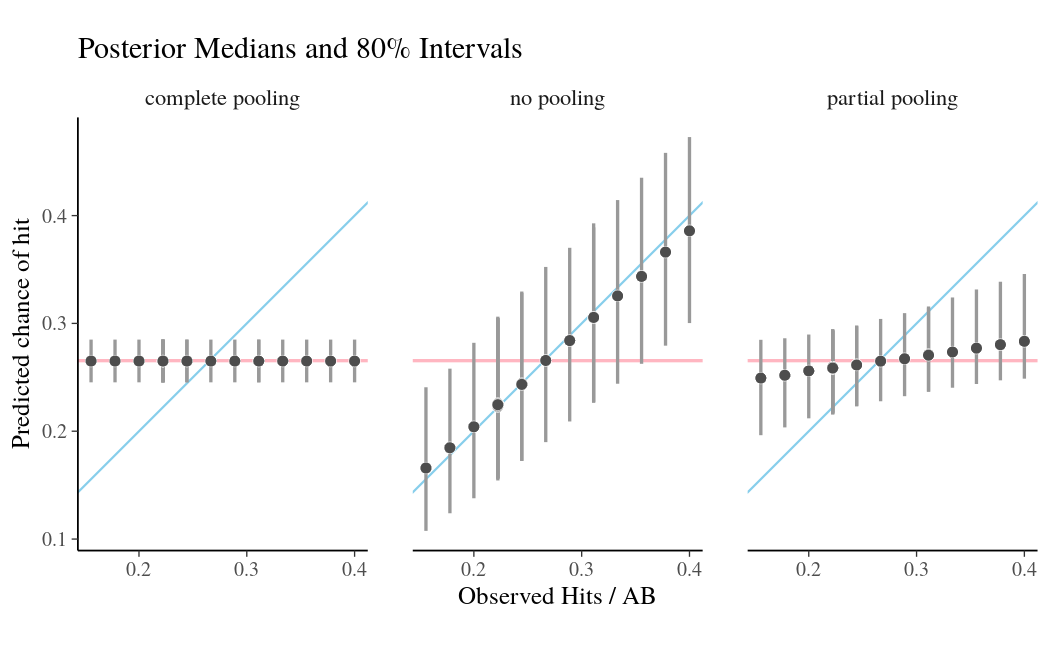
This idea is pretty simple, we’re just saying that we think the relative fitnesses should be similar across geographies: \[ \lambda_{v, g} \sim \text{Normal}(\bar{\lambda}_v, \sigma_{v}) \]
\(\lambda_{v,g}\) is the relative fitness of variant \(v\) in geography \(g\).
\(\bar{\lambda}\) is the mean relative fitness of variant \(v\) across geographies.
\(\sigma_v\) is the standard deviation in the relative fitness of \(v\).
This is called partial pooling and it allows us to share information about the relative fitness across locations.
Low-data locations can receive information about variants that they haven’t seen yet or are below detectable levels.
Automating SARS-CoV-2 variant frequency forecasts: forecasts-ncov
- We’ve developed a pipeline for:
- provisioning these sequence count data sets from both GISAID and open data,
- running the hierarchical MLR models on these data sets,
- and visualizing their results at https://nextstrain.org/sars-cov-2/forecasts/.
- This work was done with Jover Lee, James Hadfield, and Trevor Bedford.



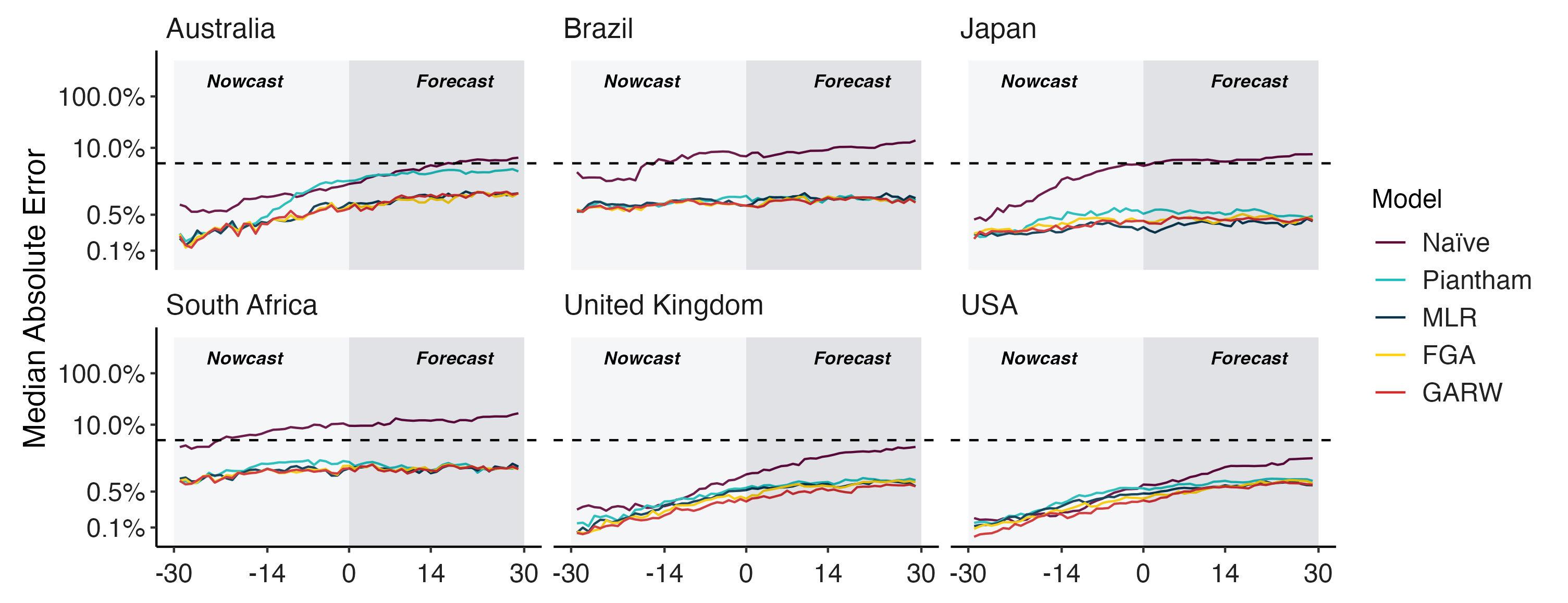
Evaluating forecasts
Fitness models provide accurate short-term forecasts of SARS-CoV-2 variant frequency

Forecasting variant frequencies is complicated!
- As we continue to develop these kinds of models, it’s essential to think about what they can do and when they work best!
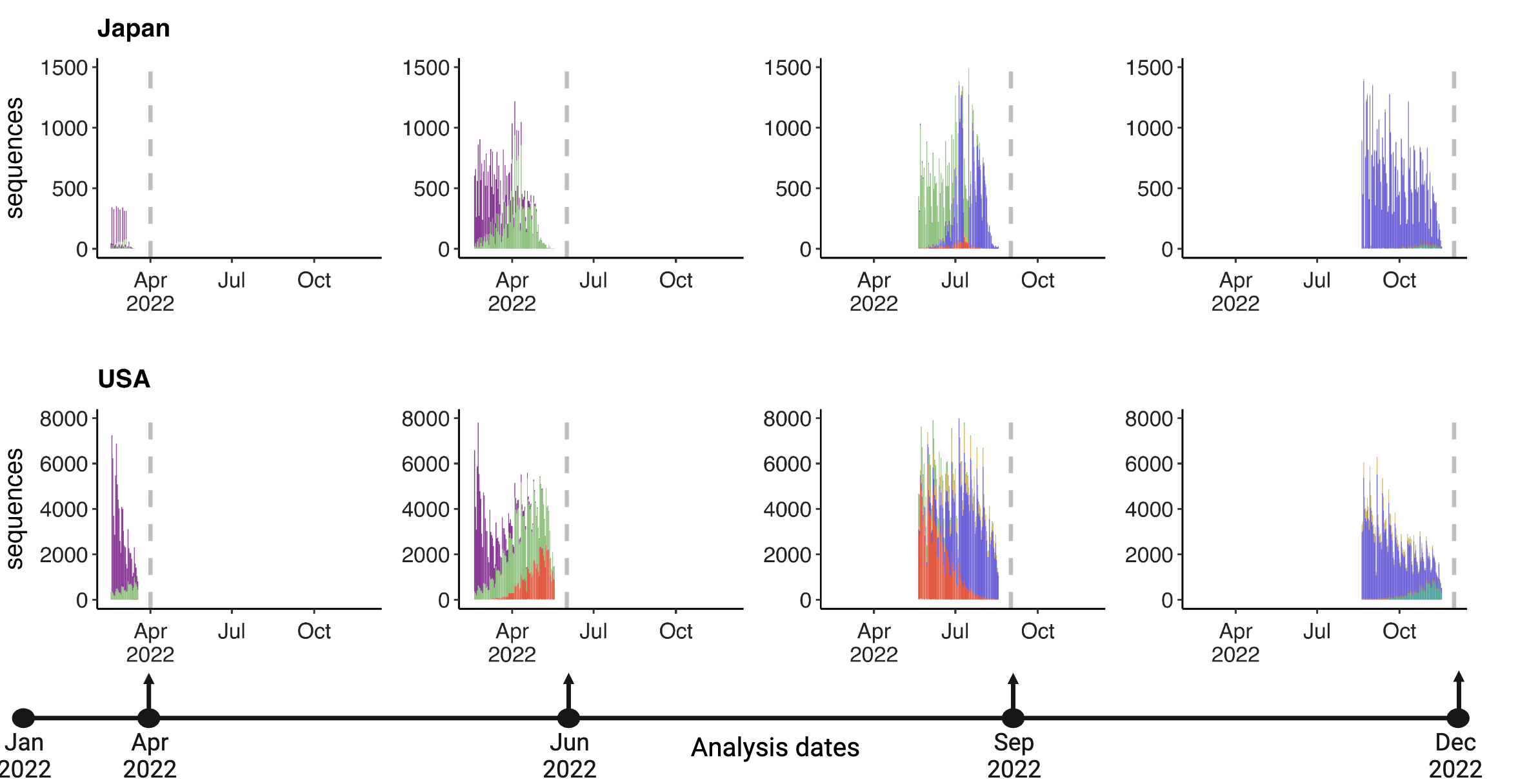
Mimicking real-time forecast environments
- As we continue to develop these kinds of models, it’s essential to think about what they can do and when they work best!
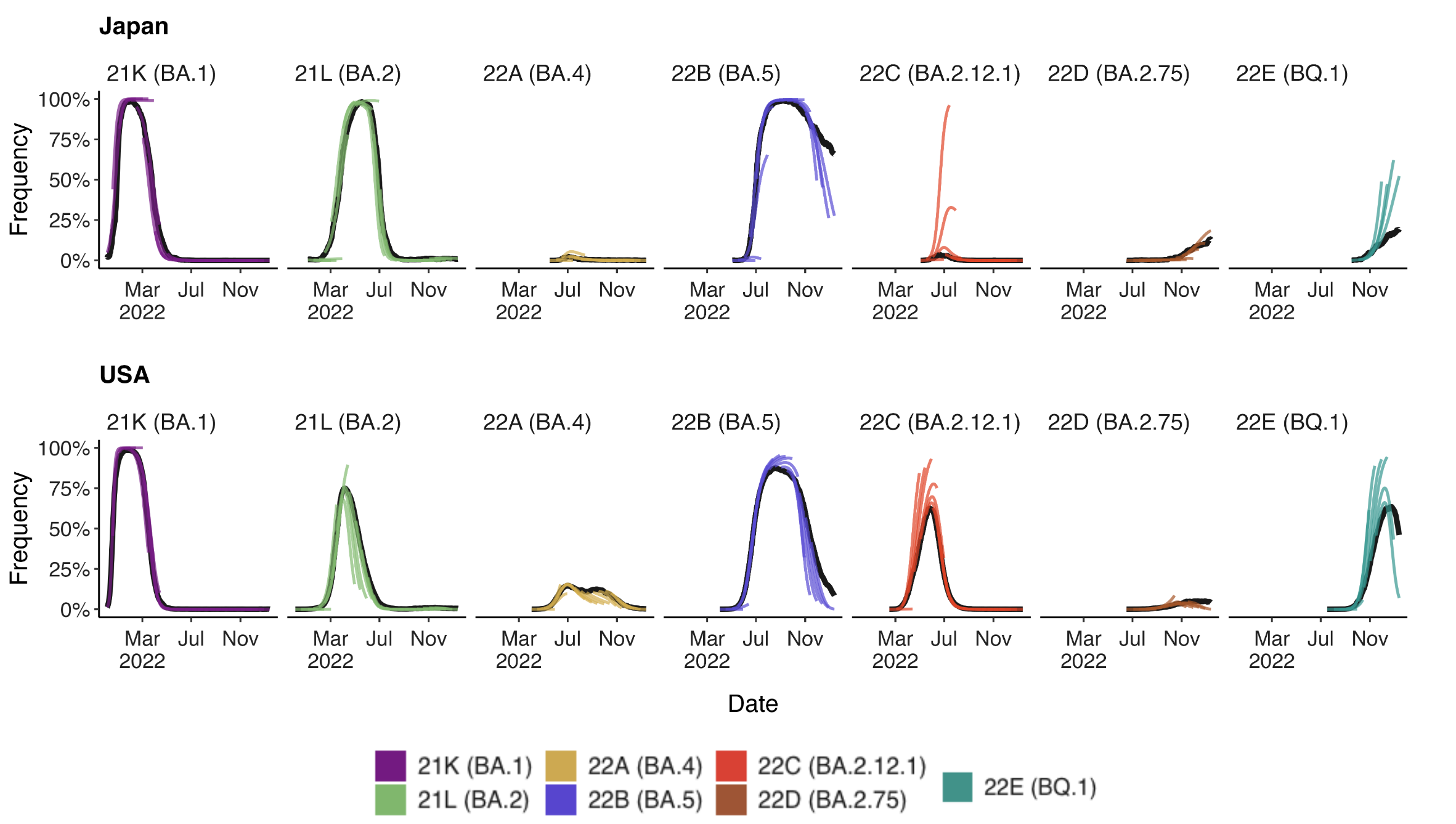
Evaluating forecasts
- We find that in general existing frequency dynamic models work well for short-term forecasts.